29-30 września mieliśmy okazję uczestniczyć w „Farmacji Jutra” – wydarzeniu łączącym uroczyste otwarcie pierwszego z budynków kompleksu Collegium Pharmaceuticum oraz Centrum Innowacyjnej Technologii Farmaceurtczneh z Konferencją pt. „NEW TRENDS in Polish and global pharmacy”.
Collegium Pharmaceuticum to przestrzeń, gdzie edukacja, badania, technologia oraz biznes przenikają się wzajemnie, wyznaczając standardy na przyszłość. Z kolei dzięki uruchomieniu CITF innowacyjne prace badawczo-rozwojowe prowadzone przez UMP i ich urynkowienie przyczynią się do podniesienia poziomu konkurencyjności, szczególnie w kontekście komercjalizacji badań naukowych i transferu wiedzy, a także budowania i utrzymywania stałych relacji ze środowiskiem gospodarczym.
Pracownicy Curtis Health Caps, w składzie: Marlena Dudek-Makuch, Natalia Palacz oraz Miłosz Regulski (Dział R&D) dostępni byli dla Odwiedzających na stoisku CHC, które znajdowało się w części wystawienniczej.
Bardzo się cieszymy, że mogliśmy być jednym ze sponsorów wydarzenia, spotkać się z Uczestnikami oraz porozmawiać o możliwościach rozwoju współczesnej farmacji.
W dniach 20-22 września br. odbywa się QUALITY TOUR – wydarzenie poświęcone najlepszym praktykom z zakresu zarządzania jakością. Główny punkt programu stanowią poparte merytoryczną prelekcją wizyty w zakładach produkcyjnych – w tym w firmie Curtis Health Caps.
W ramach tegorocznej edycji Magdalena Żarna (Kierownik Zapewnienia Jakości, Curtis Health Caps) wystąpiła z prezentacją pt. „Zarządzanie procesem kwalifikacji dostawców”. Podczas prelekcji podkreślone zostało, że dobrze zorganizowany proces zarządzania kwalifikacją dostawców jest istotnym i integralnym elementem Systemu Zarządzania Jakością. W trakcie prezentacji przedstawiony został proces zarządzania kwalifikacją dostawców oraz proces oceny ryzyka różnych kategorii rejestracyjnych produktów mający na celu określenie potrzeby wykonywania audytów dostawców.
Po prelekcji mieliśmy ogromną przyjemność gościć Uczestników QUALITY TOUR w zakładzie produkcyjnym Curtis Health Caps w Wysogotowie – 20 września odbyło się merytoryczne zwiedzanie o charakterze techniczno-jakościowym, poprowadzone przez przedstawicieli poszczególnych obszarów w firmie.Zarządzanie jakością w Curtis Health Caps, to Quality przez duże „Q” i Profesjonalizm przez duże „P.” – to jedno z podsumowań procesów dotyczących jakości. Dziękujemy!
Serdecznie dziękujemy również Uczestnikom za obecność, zaangażowanie, wymianę doświadczeń oraz inspirujące rozmowy. Zapraszamy do odwiedzenia kanałów CHC Facebook oraz LinkedIn.
Napisane przez Karolina w dniu . Opublikowano w BLOG.
Rozwój produktu leczniczego (ang. drug development) to zaplanowany, wieloetapowy proces prowadzony z zakładzie farmaceutycznym, mający na celu opracowanie i zarejestrowanie produktu leczniczego. W zależności od rodzaju produktu (np. lek oryginalny, lek generyczny, tradycyjny lek roślinny) proces ten może trwać kilka lub kilkanaście lat. Zwieńczeniem tego przedsięwzięcia jest uzyskanie Pozwolenia na dopuszczenie do obrotu, które stanowi dokument wydawany przez organ kompetentny i jest niezbędny do rozpoczęcia sprzedaży nowego produktu leczniczego.Firmy nie zawsze dysponują własnym zapleczem produkcyjnym, wówczas zlecają rozwój produktu leczniczego firmom farmaceutycznym, specjalizującym się w kontraktowym rozwoju i wytwarzaniu produktów leczniczych (CDMO, ang. Contract Development and Manufacturing Organisation).
Jakie są etapy rozwoju produktu leczniczego?
Liczba etapów, zakres prac i koszty związane z rozwojem są ściśle powiązane z rodzajem opracowywanego produktu leczniczego. Oprócz standardowych etapów, które należy zrealizować przed komercjalizacją leku, pod uwagę należy wziąć dodatkowe – ale również niezbędne – prace i koszty, np. badania kliniczne I, II oraz III fazy (w przypadku leków oryginalnych), czy badanie biorównoważności (w przypadku leku generycznego).
Standardowymi etapami rozwoju produktu leczniczego są:
Rozeznanie wstępne (analiza literatury, czystości patentowej, danych z baz rejestracyjnych)
Prace preformulacyjne
Prace i doświadczenia w skali laboratoryjnej
Optymalizacja i skalowanie procesu do wielkości pilotażowej lub produkcyjnej
Rejestracja produktu leczniczego i uzyskanie pozwolenia na dopuszczenie do obrotu
Wprowadzenie do obrotu – komercjalizacja produktu
1. Rozeznanie wstępne
Etap ten ma na celu zebranie danych mających kluczowe znaczenie w zaplanowaniu odpowiednich prac formulacyjnych. Podczas rozeznania wstępnego identyfikowane są produkty lecznicze referencyjne lub podobne pod względem postaci i składu, jaki ma mieć docelowy lek. Typowane są substancje czynne i pomocnicze, które zostaną przetestowane podczas prac rozwojowych oraz oceniane są dostępne patenty i zgłoszenia patentowe w celu identyfikacji przeszkód oraz ograniczeń, które należy uwzględnić przy rozwijaniu nowej formulacji leku.
2. Prace preformulacyjne
Etap ten w procesie rozwoju nowego produktu leczniczego jest najistotniejszy z dwóch głównych powodów: Pierwszy stanowi określenie ogólnego sposobu wytwarzania nowego produktu (materiały wyjściowe, technologia produkcji i pakowania), który wykorzystuje się do obliczania kosztów wytwarzania i na podstawie którego podejmowana jest decyzja o dalszych pracach rozwojowych;
Drugim jest wstępne wyznaczenie atrybutów jakościowych dla materiałów wyjściowych, półproduktów i produktu gotowego, na podstawie których opracowuje się plan i zakres czynności w trakcie rozwoju nowego produktu leczniczego.
Proces ten obejmuje wykonywanie prac eksperymentalnych, mających na celu opracowanie prototypów formulacji, które podlegałby dalszym modyfikacjom i badaniom w skali laboratoryjnej.
3. Prace i doświadczenia w skali laboratoryjnej
Celami tego etapu są:
opracowanie finalnego składu ilościowego i jakościowego rozwijanej formulacji leku,
ustalenie jego technologii wytwarzania wraz krytycznymi parametrami procesu,
weryfikacja (określonych w poprzedzającym etapie) zależności pomiędzy krytycznymi atrybutami materiałów wyjściowych a krytycznymi atrybutami półproduktów oraz produktu gotowego.
Oprócz realizacji prac technologicznych rozwijane są metody analityczne, które będą służyły do kontroli jakości nowego leku oraz definiowane są parametry specyfikacji produktu gotowego uwzględniające jego skład, postać i drogę podania.
4. Optymalizacja i skalowanie procesu do wielkości pilotażowej lub produkcyjnej
W ramach tego etapu wykonywane jest skalowanie procesu wytwarzania ze skali laboratoryjnej do skali półtechnicznej, a następnie do skali pilotażowej lub produkcyjnej.
Celem wytarzania leku w skali półtechnicznej jest weryfikacja i optymalizacja opracowanej technologii wytwarzania. Proces ten realizowany jest poprzez wytworzenie w warunkach produkcyjnych serii optymalizacyjnej, która analizowana jest pod kątem:
możliwości modyfikacji (i dopuszczalnego zakresu) technologii wytwarzania opracowanej w poprzednim etapie,
wpływu zmian w sposobie wytwarzania na parametry procesowe, ze szczególnym uwzględnieniem parametrów krytycznych.
Po pomyślnym wytworzeniu serii optymalizacyjnej proces ten skalowany jest do serii pilotażowej lub produkcyjnej. W przypadku standardowego procesu wytwarzania skalowanie do wielkości pilotażowej jest wystarczające do opracowania i złożenia dokumentacji do agencji rejestracyjnej. Jeśli proces jest niestandardowy (np. zawartość substancji czynnej wynosi <2% w formulacji), to należy go skalować do wielkości produkcyjnej i poddać walidacji min. 3 serie przed złożeniem wniosku o rejestrację produktu leczniczego. Biorąc pod uwagę leki generyczne należy również wytworzyć serię do badania biorównoważności, która powinna wynosić min. 100 tys. tabletek lub kapsułek, bądź stanowić min. 10% wielkości serii produkcyjnej (jeśli seria produkcyjna wynosi ponad 1 mln szt.).
Serie pilotażowe, walidacyjne i przeznaczone do badania klinicznego poddawane są również badaniom stabilności zgodnie z odpowiednimi wytycznymi.
W wyniku przeprowadzonych prac rozwojowych opracowywana jest dokumentacja jakościowa dotycząca substancji czynnej oraz produktu leczniczego. Prezentowany jest finalny skład leku, wyniki prac rozwojowych, opis sposobu wytwarzania leku i kontroli tego procesu, dane na temat substancji pomocniczych, specyfikacja produktu gotowego, opis metod analitycznych, opis materiałów opakowaniowych oraz wyniki badań stabilności. Dokumentacja prezentowana jest w formie Modułu 3 CTD, który stanowi niezbędną część całej dokumentacji składanej wraz z wnioskiem do agencji rejestracyjnej.
6. Rejestracja produktu leczniczego i uzyskanie pozwolenia na dopuszczenie do obrotu
Produkty lecznicze można rejestrować w wybranym państwie członkowskim UE, kilku państwach UE lub w całej UE – o wyborze drogi rejestracji decyduje wnioskodawca składający dokumentację.
Dokumentacja rejestracyjna złożona do agencji podlega następnie rygorystycznej ocenie pod kątem jakości, bezpieczeństwa i skuteczności produktu leczniczego. Prezentowane dane są weryfikowane pod względem zgodności z obowiązującymi wymaganiami prawnymi, a w przypadku braków lub niezgodności z przepisami, agencja rejestracyjna przesyła do wnioskodawcy wezwanie do odpowiedzi na pytania oraz usunięcia tych niezgodności. Po pomyślnej ocenie dokumentacji wydawane jest Pozwolenie na dopuszczenie do obrotu ważne przez 5 lat, które następnie może zostać przedłużone bezterminowo.
7. Wprowadzenie do obrotu – komercjalizacja produktu
Pozwolenie na dopuszczenie do obrotu jest dokumentem urzędowym wieńczącym rozwój nowego produktu leczniczego. Po jego uzyskaniu podmiot odpowiedzialny we współpracy z wytwórcą jest uprawniony do wytwarzania leku w skali produkcyjnej zgodnie z zatwierdzoną dokumentacją oraz do jego komercjalizacji poprzez sprzedaż na zarejestrowanych rynkach.
Podsumowanie
Rozwój nowego produktu leczniczego to proces skomplikowany, wieloetapowy i bardzo kosztowny. W zależności od kategorii produktu może on trwać kilka lat (np. leki generyczne lub o ugruntowanym zastosowaniu medycznym) lub kilkanaście (leki oryginalne wymagające pełnych badań klinicznych). O kosztach rozwoju leku decyduje nie tylko jego kategoria i wybór drogi rejestracji, ale również wiele innych czynników, tj. koszty materiałów wyjściowych, koszty i sposób wytwarzania, dostępny park maszynowy oraz inwestycje wymagające zakupu nowych urządzeń produkcyjnych lub laboratoryjnych, koszty walidacji wytwarzania, koszty badań przedklinicznych i klinicznych.
W związku z powyższym podjęcie decyzji o rozwoju produktu leczniczego powinno być poprzedzone szczegółową analizą korzyści ekonomicznych w stosunku do ryzyka związanego z rozwojem formulacji o nieodpowiedniej jakości, ryzyka rejestracyjnego oraz rachunku potencjalnych kosztów do poniesienia przez komercjalizacją.
Właściwie przeprowadzona ocena korzyści i zagrożeń związanych rozwojem produktu leczniczego pozwala zminimalizować ryzyko niepowodzenia przedsięwzięcia.
W dniach 29-30 września 2022 r. odbędzie się wydarzeniu łączące uroczyste otwarcie pierwszego z budynków kompleksu Collegium Pharmaceuticum oraz Centrum Innowacyjnej Technologii Farmaceutycznej (CITF) z Międzynarodową Konferencją Naukowo-Szkoleniową. Nasi Pracownicy będą obecni na stoisku wystawienniczym w nowo otwartych przestrzeniach Wydziału Farmaceutycznego.
Szczegóły wydarzenia znajdują się TUTAJ.
Od początku pandemii COVID-19 w wielu badaniach populacyjnych i przekrojowych wykazano związek między niskim stężeniem aktywnego metabolitu witaminy D (25-hydroksywitaminy D; 25(OH)D w surowicy a ryzykiem zachorowania, ciężkiego przebiegu oraz zgonu u pacjentów z COVID-19 [1]. W badaniu Mercola i wsp. pacjenci ze stężeniem 25(OH)D w surowicy > 30 ng/ml mieli łagodny przebieg choroby, podczas gdy u pacjentów ze stężeniem 25(OH)D w surowicy poniżej 30 ng/ml COVID-19 wskaźniki śmiertelności były zdecydowanie wyższe [2]. W innych badaniach wykazano, że większość hospitalizowanych pacjentów z COVID-19 miała niedobór witaminy D, co wiązało się z 3,79-krotnym wzrostem ryzyka ciężkiego przebiegu COVID-19 i 4,07-krotnym wzrostem ryzyka zgonu [3–5].
Jest to związane m.in. z wpływem witaminy D na modulowanie zarówno wrodzonej, jak i nabytej odpowiedzi immunologicznej, w tym ze zmniejszeniem aktywności neutrofili, tłumieniem nadmiernej aktywności limfocytów Th1, co zapobiega rozwojowi burzy cytokinowej [1], a także bezpośrednim pozytywnym wpływem 25(OH)D na ekspresję ACE2, receptora na powierzchni komórek gospodarza dla SARS-CoV-2 [6-8].
Obniżenie ryzyka infekcji SARS-COV-2
W najnowszym randomizowanym badaniu klinicznym oceniano skuteczność witaminy D w zapobieganiu zakażeniu SARS-CoV-2 u osób wysoko narażonych oraz jej wpływ na ciężkość przebiegu choroby. Badanie przeprowadzono na pracownikach pierwszej linii opieki zdrowotnej (pielęgniarki, lekarze), którzy opiekowali się chorymi na COVID-19, jeszcze przed wprowadzeniem szczepionki. Uczestnicy zostali losowo przydzieleni do grupy otrzymującej co dziennie 4 000 IU witaminy D (VDG; n = 94) lub placebo (PG; n = 98) przez okres 30 dni. W trakcie badania wykonywano testy RT-PCR w kierunku infekcji COVID-19, mierzono poziom przeciwciał IgG przeciwko SARS-Cov-2, a także oznaczano poziom 25(OH)D w osoczu [9].
Badanie wykazało, że suplementacja witaminy D spowodowała istotny wzrost poziomu 25(OH)D, odsetek infekcji SARS-CoV-2 był zdecydowanie niższy w grupie VDG niż w PG (6,4 vs 24,5%, p <0,001), a ryzyko zakażenia w grupie interwencyjnej było niższe o 77% (RR: 0,23; 95% CI: 0,09–0,55). Zastosowana terapia była bardzo bezpieczna, nie zidentyfikowano żadnych istotnych zdarzeń niepożądanych [9].
Wyniki tego badania jednoznacznie wskazują, że witamina D chroni przed zakażeniem SARS-CoV-2, nawet osoby wysoko narażone [9].
Skrócenie czasu powrotu do zdrowia
W innym badaniu oceniono wpływ codziennej doustnej suplementacji witaminy D3 na poprawę objawów i innych parametrów klinicznych u pacjentów z infekcją COVID-19. W badaniu wzięło udział 69 dorosłych pacjentów z dodatnim wynikiem RT-PCR w kierunku SARS-CoV-2, hospitalizowanych z powodu łagodnej lub umiarkowanej choroby COVID-19. Pacjenci zostali losowo przydzieleni do grupy otrzymującej doustnie przez 2 tygodnie witaminę D w dawce 5000 IU (n = 36) lub 1000 IU (n = 33). W trakcie badania oceniano poziom 25(OH)D, parametry morfologiczne krwi, profile nerkowy i wątrobowy, markery stanu zapalnego a także czas do ustąpienia objawów choroby, w tym gorączki, bólu głowy, duszności, kaszlu, bólu gardła, zmęczenia, utraty węchu i smaku. Badanie wykazało, że w grupie przyjmującej 5000 IU u chorych znacznie szybciej ustępowały objawy choroby niż w grupie 1000 IU (6,2±0,8 w porównaniu do 9,1±0,8 dni), a także szybciej powracał smak (11,4±1,0 vs 16,9±1,7; p = 0,035) i zapach (11,2±1,1 vs 16,3±1,7; p = 0,14) [10].
Badanie to dowodzi, że codzienna suplementacja witaminy D3 w dawce 5000 IU w trakcie infekcji COVID-19 skraca czas powrotu do zdrowia i może być zalecana jako terapia uzupełniająca.
Piśmiennictwo
Dudek-Makuch M. Suplementacja witaminy D w prewencji zakażeniem SARS-COV-2 i łagodzenia przebiegu COVID-19. https://chc.com.pl/suplementacja-witaminy-d-w-prewencji-zakazeniem-sars-cov-2-i-lagodzenia-przebiegu-covid-19/
Mercola J, Grant WB, Wagner CL. Evidence Regarding Vitamin D and Risk of COVID-19 and Its Severity. Nutrients. 2020 Oct 31;12(11):3361. doi: 10.3390/nu12113361. PMID: 33142828; PMCID: PMC7692080.4 ].
Karonova TL, Andreeva AT, Golovatuk KA, Bykova ES, Simanenkova AV, Vashukova MA, Grant WB, Shlyakhto EV. Low 25(OH)D Level Is Associated with Severe Course and Poor Prognosis in COVID-19. Nutrients. 2021 Aug 29;13(9):3021. doi: 10.3390/nu13093021. PMID: 34578898; PMCID: PMC8468115.
Karonova TL, Kudryavtsev IV, Golovatyuk KA, Aquino AD, Kalinina OV, Chernikova AT, Zaikova EK, Lebedev DA, Bykova ES, Golovkin AS, Shlyakhto EV. Vitamin D Status and Immune Response in Hospitalized Patients with Moderate and Severe COVID-19. Pharmaceuticals (Basel). 2022 Mar 2;15(3):305. doi: 10.3390/ph15030305. PMID: 35337103; PMCID: PMC8955127.
Carpagnano GE, Di Lecce V, Quaranta VN, Zito A, Buonamico E, Capozza E, Palumbo A, Di Gioia G, Valerio VN, Resta O. Vitamin D deficiency as a predictor of poor prognosis in patients with acute respiratory failure due to COVID-19. J Endocrinol Invest. 2021 Apr;44(4):765-771. doi: 10.1007/s40618-020-01370-x. Epub 2020 Aug 9. PMID: 32772324; PMCID: PMC7415009.
Agraz-Cibrian JM, Giraldo DM, Urcuqui-Inchima S. 1,25-Dihydroxyvitamin D3 induces formation of neutrophil extracellular trap-like structures and modulates the transcription of genes whose products are neutrophil extracellular trap-associated proteins: A pilot study. Steroids. 2019 Jan;141:14-22. doi: 10.1016/j.steroids.2018.11.001. Epub 2018 Nov 7. PMID: 30414422.
Bouillon R, Marcocci C, Carmeliet G, Bikle D, White JH, Dawson-Hughes B, Lips P, Munns CF, Lazaretti-Castro M, Giustina A, Bilezikian J. Skeletal and Extraskeletal Actions of Vitamin D: Current Evidence and Outstanding Questions. Endocr Rev. 2019 Aug 1;40(4):1109-1151. doi: 10.1210/er.2018-00126. PMID: 30321335; PMCID: PMC6626501.
Xu J, Yang J, Chen J, Luo Q, Zhang Q, Zhang H. Vitamin D alleviates lipopolysaccharide‑induced acute lung injury via regulation of the renin‑angiotensin system. Mol Med Rep. 2017 Nov;16(5):7432-7438. doi: 10.3892/mmr.2017.7546. Epub 2017 Sep 20. PMID: 28944831; PMCID: PMC5865875.
Villasis-Keever MA, López-Alarcón MG, Miranda-Novales G, Zurita-Cruz JN, Barrada-Vázquez AS, González-Ibarra J, Martínez-Reyes M, Grajales-Muñiz C, Santacruz-Tinoco CE, Martínez-Miguel B, Maldonado-Hernández J, Cifuentes-González Y, Klünder-Klünder M, Garduño-Espinosa J, López-Martínez B, Parra-Ortega I. Efficacy and Safety of Vitamin D Supplementation to Prevent COVID-19 in Frontline Healthcare Workers. A Randomized Clinical Trial. Arch Med Res. 2022 Jun;53(4):423-430. doi: 10.1016/j.arcmed.2022.04.003. Epub 2022 Apr 18. PMID: 35487792; PMCID: PMC9013626.
Sabico S, Enani MA, Sheshah E, Aljohani NJ, Aldisi DA, Alotaibi NH, Alshingetti N, Alomar SY, Alnaami AM, Amer OE, Hussain SD, Al-Daghri NM. Effects of a 2-Week 5000 IU versus 1000 IU Vitamin D3 Supplementation on Recovery of Symptoms in Patients with Mild to Moderate Covid-19: A Randomized Clinical Trial. Nutrients. 2021 Jun 24;13(7):2170. doi: 10.3390/nu13072170. PMID: 34202578; PMCID: PMC8308273.
Nota biograficzna
Dr n. farm. Marlena Dudek-Makuch, Ekspert ds. Rozwoju w Curtis Health Caps, Wysogotowo.
Posiada 20-letnie doświadczenie w zakresie badań fitochemicznych i biologicznych oraz informacji naukowej (adiunkt w Katedrze i Zakładzie Farmakognozji Uniwersytetu Medycznego w Poznaniu). Autorka prac eksperymentalnych i poglądowych z zakresu izolacji i identyfikacji związków pochodzenia roślinnego oraz oceny ich aktywności biologicznej. Od 2015 roku prowadzi zajęcia na studiach podyplomowych „Zioła w praktyce i terapii”.
Obecnie pracuje w CHC w Pionie R&D, Dziale Regulacji. Odpowiada m.in. za opracowanie raportów Eksperta (raport kliniczny, nieklinicznych) dla produktów leczniczych, raportu klinicznego do zmian kategorii dostępności produktu leczniczego (switch OTC), oceny klinicznej dla wyrobów medycznych oraz prowadzenie działań w obszarze nadzoru nad bezpieczeństwem wyrobów medycznych, a także za ocenę bezpieczeństwa surowców roślinnych stosowanych w produktach leczniczych, wyrobach medycznych i suplementach diety.
Jest członkiem Polskiego Komitetu Zielarskiego.
Miło nam poinformować, że 29.09.2022 r. podczas Konferencji Zakupowej Procurement Angels, swoją prelekcję wygłosi Kierownik Działu Zakupów CHC, Anna Frąckowiak. Zaproszenie na wykład można obejrzeć na naszym profilu na Linkedin. Trzymamy kciuki za Anię, ponieważ uczestnicy konferencji wybiorą najlepszą prelekcję roku 2022.
Szczegóły Konferencji Zakupowej Procurement Angels znajdują się tutaj.
W ostatnim czasie wiele się mówi o wielokierunkowym, prozdrowotnym wpływie witaminy D na nasz organizm, badania naukowe potwierdzają nie tylko jej korzystny wpływ na zdrowie naszych kości, ale również na poprawę odporności, funkcjonowanie układu krwionośnego, regulację metabolizmu węglowodanów, czy wspieranie zdrowia układu nerwowego [1]. Wiele badań potwierdza jednak ogromny niedobór witaminy D zarówno wśród dzieci, młodzieży, osób dorosłych i starszych, a aktualne rekomendacje towarzystw naukowych podkreślą konieczność jej suplementacji na obszarach leżących powyżej 33° szerokości geograficznej (cała Europa) w okresie od października do kwietnia [2].
Pojawiają się jednak głosy, że tak masowa suplementacja nie jest wskazana oraz że może być ona szkodliwa a nawet toksyczna dla organizmu. Jaka jest zatem prawda, czy witamina D stosowana w zalecanej ilości przez dłuższy okres czasu jest bezpieczna? Czy możliwe jest jej przedawkowanie, jakie są jego skutki i kiedy może do niego dojść?
Aby dobrze zrozumieć podstawy szerokiego bezpieczeństwa witaminy D konieczne jest zapoznanie z podstawowymi wiadomościami dotyczącymi jej metabolizmu i mechanizmów autoregulacji stężenia aktywnych metabolitów w organizmie.
Metabolizm witaminy D
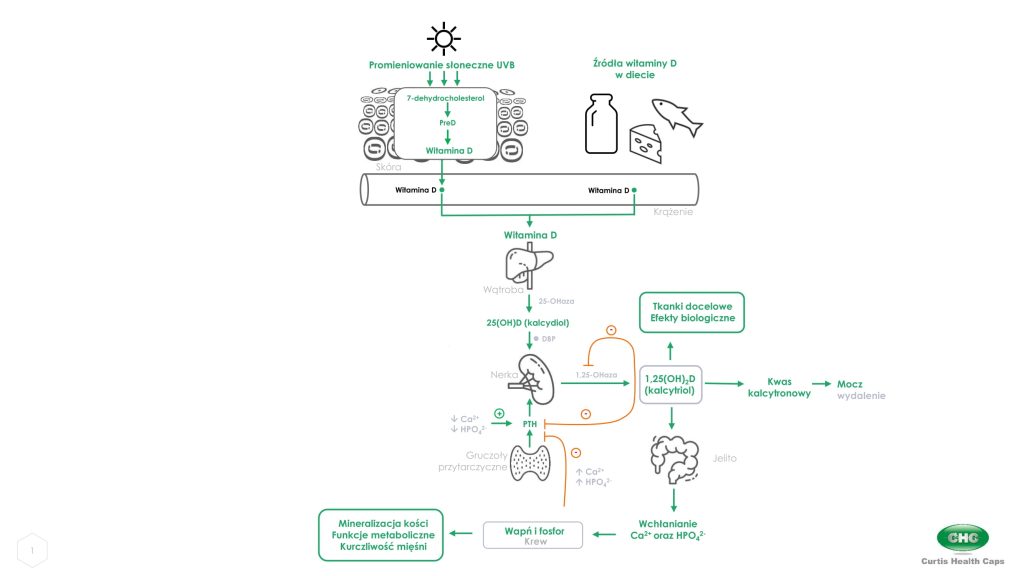
Witamina D otrzymywana po ekspozycji słonecznej, przyjmowana z żywnością lub suplementami diety jest biologicznie nieaktywna i musi przejść aktywację w dwóch kolejnych enzymatycznych reakcjach hydroksylacji zachodzących w wątrobie i nerkach (ryc. 1).
Endogennie witamina D (cholekalcyferol) jest fotosyntetyzowana w skórze. Obecny w skórze 7-dehydrocholesterol (prowitamina D) po ekspozycji na promieniowanie ultrafioletowe B (UVB 290-315 nm) ulega w naskórku przekształceniu do formy prekalcyferolu (PreD), a następnie do witaminy D (ryc. 1). Produkcja witaminy D w skórze wynika z zakresu i jakości promieniowania UVB docierającego do skóry właściwej, a także z dostępności 7-dehydrocholesterolu i właściwości skóry.
Dietetyczna witamina D jest wchłaniana w jelicie cienkim wraz z innymi tłuszczami pokarmowymi [3]. W wątrobie prekalcyferol jest szybko hydroksylowany przez 25-hydroksylazę do nieaktywnego 25-hydroksycholekalcyferolu (25(OH)D, kalcydiolu) [4], który po związaniu z białkiem (ang. Vitamin D-Binding Protein; DBP), jest z krwią transportowany do nerek, gdzie ulega hydroksylacji przez 1,25-hydroksylazę do aktywnej formy 1,25-dihydroksycholekalcyferolu (1,25(OH)2D, kalcytriolu). Średni czas obecności 25(OH)D w osoczu wynosi około 3 tygodni i to właśnie sprawia, że stężenie 25(OH)D w surowicy krwi jest wskaźnikiem statusu witaminy D w organizmie.
Poziom aktywnej formy witaminy D (1,25(OH)2D) jest bardzo ściśle regulowany przez poziom wapnia i fosforanów we krwi za pośrednictwem hormonu przytarczyc (PTH) i czynnika wzrostu fibroblastów 23 (FGF-23) [5].
Regulacja metabolizmu witaminy D
Stężenie witaminy D i 25(OH)D w surowicy zależy od pory roku i spożycia witaminy D [5], natomiast stężenie aktywnego metabolitu 1,25(OH)2D w surowicy jest niezmienne (16-65 pg/mL) i niezależne od ekspozycji na światło słoneczne czy suplementację. Przy odpowiednim poziomie 25(OH)D w organizmie, kluczową rolę w metabolizowaniu 25(OH)D do biologicznie aktywnego metabolitu – 1,25(OH)2D odgrywają nerki.
W przypadku hipokalcemii (niskiego poziomu wapnia we krwi) dochodzi do zwiększonego wydzielania PTH oraz zwiększenia aktywności i szybkości nerkowej hydroksylacji oraz tworzenia aktywnego metabolitu witaminy D (1,25(OH)2D), który następnie zwiększa reabsorpcję (wchłanianie zwrotne) wapnia z kanalików podnosząc jego stężenie we krwi (ryc.1).
Z kolei wysoki poziom 1,25(OH)2D wpływa również na nerkowy metabolizm 25(OH)D (kalcydiol), zmniejszając aktywność 25-hydrolaz i zwiększając metabolizm kalcydiolu do 24,25(OH)2D (nieaktywny metabolit). Prowadzi to do degradacji zarówno 25(OH)D, jak i 1,25(OH)2D do rozpuszczalnych w wodzie nieaktywnych metabolitów oraz wydalenia ich z organizmu [6]. To z kolei prowadzi do obniżenia poziomu aktywnych metabolitów witaminy D w organizmie i normalizacji poziomy wapnia we krwi.
Ryc. 1. Metabolizm witaminy D i mechanizmy regulujące jej poziom w organizmie. Grafika własna na podstawie Holick 2006 [6].
Bezpieczeństwo
Witamina D jest uważana za ogólnie bezpieczną, gdyż jej poziom w organizmie podlega regulacji wewnętrznej i utrzymywany jest w zakresie stężeń fizjologicznych. Suplementacja witaminy D przez pacjentów z niedoborem tej substancji jest na ogół bardzo dobrze tolerowana, a toksyczne poziomy są rzadko obserwowane w praktyce klinicznej [7].
Zgodnie z najnowszymi standardami dawkowania witaminy D opracowanymi przez EFSA (European Food Safety Authority) oraz IOM (Institute of Medicine) w USA, dzienna bezpieczna dawka dla osób dorosłych wynosi 4000 IU/dzień, można ją zwiększyć do 10 000 IU w przypadku zaburzeń wchłaniania lub patologicznej otyłości (po konsultacji lekarskiej) [8]. Zgodnie z rekomendacjami GIS w Polsce witamina D w dawce 4000 IU jest dostępna jako suplement diety (dla osób zdrowych powyżej 75 roku życia) [9], zaś wyższe dawki dostępne są w postaci produktów leczniczych.
Górna granica bezpieczeństwa oparta jest na wyznaczeniu korelacji między poziomem 25(OH)D w surowicy krwi a ryzykiem wystąpienia hiperkalcemii (zbyt dużego poziomu wapnia we krwi) [10-13]. Wykazano, że dawka 95 μg/dzień (3800 IU/dzień) stosowana przewlekle jest najniższą dawką, która może zwiększyć ryzyko hiperkalcemii u zdrowych dorosłych, jednakże podwyższony poziom wapnia w surowicy krwi stwierdzono zaledwie u sześciu osób spożywających 95 μg/dzień przez 3 miesiące [14]. W innych badaniach porównano bezpieczeństwo 5-miesiacznej suplementacji witaminą D w dawkach 100 μg/dzień (4000 IU) i 25 μg/dzień (1000 IU). Maksymalne poziomy 25(OH)D w surowicy wynosiły odpowiednio 48 ng/mL (120 nmol/L) i 40 ng/mL (100 nmol/L), ani stężenie wapnia w surowicy, ani stosunek wapnia do kreatyniny w stolcu nie zmieniły się znacząco podczas badania [12]. W niedawnym badaniu obejmującym 373 starsze zdrowe osoby z niedoborem witaminy D, którym podawano 400, 4000 i 10 000 IU dziennie przez 3 lata, wykazano, że hiperkalcemia (całkowity wapń w surowicy > 2,55 mmol/L) wystąpiła jedynie u 0, 3 i 9% (odpowiednio w grupach 400, 4000 i 10 000 IU/dzień), zaś hiperkalciurię (24-godzinne wydalanie wapnia z moczem powyżej 7,5 mmol/dobę) wykryto u 17, 22 i 31% (w analogicznych grupach) [15].
Dawki toksyczne
Ze względu na szeroki indeks terapeutyczny (stosunek dawki leku powodującej objawy zatrucia do dawki skutecznej leczniczo), przedawkowanie witaminy D występuje rzadko i zdarza się jedynie przy stosowaniu zbyt dużych dawek. Przyjmowanie 30 000 IU witaminy D dziennie przez ponad 3 miesiące jest uważane za dawkę toksyczną. Przy takiej suplementacji, zgodnie z raportem IOM opublikowanym 30 listopada 2010 roku stężenie 25(OH)D w surowicy może osiągnąć 200 ng/mL, co niesie za sobą duże ryzyko hiperkalcemii i zaburzeń metabolizmu wapnia [16].
Przyczyny przedawkowania
Przedawkowanie witaminy D prowadzące do jawnej hiperkalcemii jest rzadkie i może wynikać z endogennej nadprodukcji aktywnego metabolitu 1,25(OH)2D poprzez 1-α-hydroksylację w nieprawidłowych makrofagach, co spotyka się w sarkoidozie (przewlekła choroba śródmiąższowa płuc, rzadziej obejmująca inne narządy), lub poprzez uwalnianie witaminy D z zapasów tkanki tłuszczowej w przypadku szybkiej utraty masy tłuszczowej. Nadmiar witaminy D wiąże się ze zwiększonym jelitowym wchłanianiem wapnia i resorpcją kości (wchłanianiem składników mineralnych w tym wapnia przez kości) [17].
W literaturze naukowej opisywane są również przypadki przedawkowania witaminy D po zastosowaniu bardzo dużych dawek, rzadko są one spowodowane błędami związanymi z wytwarzaniem preparatów, częściej dochodzi do omyłkowego (błędne dawkowanie) lub świadomego (kierowanie się informacjami z niesprawdzonych źródeł) przyjmowania preparatów witaminy D w ekstremalnie wysokich dawkach.
Wysokie dawki są szczególnie niebezpieczne dla niemowląt i małych dzieci, ze względu na słabo rozwinięte mechanizmy samoregulacji. W piśmiennictwie naukowym opisane są przypadki niemowląt, które przyjmowały dawki witaminy D dawki nawet 25-krotnie wyższe od zalecanej, przez okres kilku miesięcy, co w efekcie prowadziło do hiperwitaminozy D, ciężkiej hiperkalcemii objawiających się znacznym odwodnieniem, wymiotami, zaparciami, sennością i utratą masy ciała [18-19].
Rocha i Santos opisali przypadek 19-letniego mężczyzny, który w celu uzyskania zwiększonego przyrostu masy mięśniowej stosował parenteralny (dożylnie podawany) preparat witamin A, D i E przeznaczony do użytku weterynaryjnego (5 000 000 IU witaminy D w fiolce o pojemności 100 ml). W ciągu roku przyjął 300 ml preparatu (15 000 000 IU/rok), co w konsekwencji doprowadziło do spadku masy ciała, wystąpienia nudności i wymiotów, a także hiperwitaminozy (stężenie 25(OH)D w surowicy wynosiło 150 ng/mL) i hiperkalcemii [20].
Niekiedy lekarze decydują się na leczenie pacjentów dużymi dawkami witaminy D, które przekraczające standardowe zalecenia. Opisano przypadki pacjentów, którym przepisano wysokie dawki witaminy D na różne dolegliwości. Ustalono, że pacjenci przyjmowali 2 220 000-60 000 000 IU witaminy D przez okres 4-7 tygodni z powodu różnych schorzeń. Stosowanie megadawek spowodowało wystąpienie hiperwitaminozy (25(OH)D w zakresie 164-1161 ng/mL) oraz hipercalcemii (stężeniem wapnia w surowicy w granicach 11,0-15,7 mg/dL), co klinicznie objawiało się nadmierną sennością, nudnościami i wymiotami, poliurią oraz dysfunkcja nerek [21-24].
Podsumowanie
Witamina D stosowana w dawkach zgodnych z rekomendacjami jest bezpieczna.
Przedawkowanie witaminy D występuje bardzo rzadko, ze względu na fakt, że jest ona substancją fizjologiczną, jej odpowiedni poziom jest regulowany przez organizm.
W piśmiennictwie opisywano przypadki przedawkowania witaminy D, które były spowodowane podawaniem bardzo dużych dawek (>30 000/dobę) przez długi okres czasu.
Przedawkowanie powoduje hiperwitaminozę D, hiperkalcemię, które prowadzą do bólów brzucha, wymiotów, odwodnienia, braku apetytu, zmęczenia i wielomoczu. Żaden przypadek przedawkowania nie zakończył się śmiercią pacjenta.
Piśmiennictwo
Christakos S, Li S, De La Cruz J, Bikle DD. New developments in our understanding of vitamin metabolism, action and treatment. Metabolism. 2019 Sep;98:112-120. Doi: 10.1016/j.metabol.2019.06.010. Epub 2019 Jun 19. PMID: 31226354; PMCID: PMC6814307.
Rusińska A, Płudowski P, Walczak M, Borszewska-Kornacka MK, Bossowski A, Chlebna-Sokół D, Czech-Kowalska J, Dobrzańska A, Franek E, Helwich E, Jackowska T, Kalin M, Konstantynowicz J, Książyk J, Lewiński A, Łukaszkiewicz J, Marcinowska-Suchowierska E, Mazur A, Michałus I, Peregud-Pogorzelski J, Romanowska H, Ruchała M, Socha P, Szalecki M, Wielgoś M, Zwolińska D, Zygmunt A. Zasady suplementacji i leczenia witaminą D – nowelizacja 2018 r. Standardy Medyczne/Pediatria. 2018;15:531-559.
Silva MC, Furlanetto TW. Intestinal absorption of vitamin D: a systematic review. Nutr Rev 2018; 76: 60–76
Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr. 2008 Aug;88(2):582S-586S.
Norman AW: Sunlight, season, skin pigmentation, vitamin D, and 25-hydroxyvitamin D: Integral components of the vitamin D endocrine system. Am J Clin Nutr 1998; 67: 1108–1110.
Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006 Mar;81(3):353-73. Doi: 10.4065/81.3.353. PMID: 16529140.
Nowson CA. Prevention of fractures in older people with calcium and vitamin D. Nutrients. 2010;2:975-84.
Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, Durazo-Arvizu RA, Gallagher JC, Gallo RL, Jones G, Kovacs CS, Mayne ST, Rosen CJ, Shapses SA. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96:53-58.
Uchwała GIS nr 1/2021 Zespołu ds. Suplementów Diet zmieniająca Uchwałę nr 4/2019 w sprawie wyrażania opinii dotyczącej maksymalnej dawki witaminy D w zalecanej dziennej porcji w suplementach diety.
Gallagher JC, Sai A, Templin T 2nd, Smith L. Dose response to vitamin D supplementation in postmenopausal women: a randomized trial. Ann Intern Med. 2012 Mar 20;156(6):425-37. Erratum in: Ann Intern Med. 2012 May 1;156(9):672.
Vieth R. Vitamin D supplementation, 25-hydroxyvitamin D concentrations, and safety. Am J Clin Nutr. 1999 May;69(5):842-56.
Vieth R, Chan PC, MacFarlane GD. Efficacy and safety of vitamin D3 intake exceeding the lowest observed adverse effect level. Am J Clin Nutr. 2001 Feb;73(2):288-94.
Bischoff-Ferrari HA, Shao A, Dawson-Hughes B, Hathcock J, Giovannucci E, Willett WC. Benefit-risk assessment of vitamin D supplementation. Osteoporos Int. 2010 Jul;21(7):1121-32.
Narang NK, Gupta RC, Jain MK. Role of vitamin D in pulmonary tuberculosis. J Assoc Physicians India. 1984 Feb;32(2):185-8.
Billington EO, Burt LA, Rose MS, Davison EM, Gaudet S, Kan M, Boyd SK, Hanley DA. Safety of High-Dose Vitamin D Supplementation: Secondary Analysis of a Randomized Controlled Trial. J Clin Endocrinol Metab. 2020 Apr 1;105(4):dgz212. Erratum in: J Clin Endocrinol Metab. 2021 Mar 25;106(4):e1932.
Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr. 2008 Aug;88(2):582S-586S. doi: 10.1093/ajcn/88.2.582S. PMID: 18689406.
Rizzoli R, Stoermann C, Ammann P, Bonjour JP. Hypercalcemia and hyperosteolysis in vitamin D intoxication: effects of clodronate therapy. Bone. 1994 Mar-Apr;15(2):193-8.
Ketha H, Wadams H, Lteif A, Singh RJ. Iatrogenic vitamin D toxicity in an infant–a case report and review of literature. J Steroid Biochem Mol Biol. 2015 Apr;148:14-8.
Bilbao NA. Vitamin D Toxicity in Young Breastfed Infants: Report of 2 Cases. Glob Pediatr Health. 2017 Sep 19;4:2333794X17731695.
Rocha PN, Santos CS, Avila MO, Neves CL, Bahiense-Oliveira M. Hypercalcemia and acute kidney injury caused by abuse of a parenteral veterinary compound containing vitamins A, D, and E. J Bras Nefrol. 2011 Dec;33(4):467-71
Kaur P, Mishra SK, Mithal A. Vitamin D toxicity resulting from overzealous correction of vitamin D deficiency. Clin Endocrinol (Oxf). 2015 Sep;83(3):327-31.
Koul PA, Ahmad SH, Ahmad F, Jan RA, Shah SU, Khan UH. Vitamin d toxicity in adults: a case series from an area with endemic hypovitaminosis d. Oman Med J. 2011 May;26(3):201-4.
Pandita KK, Razdan S, Kudyar RP, Beigh A, Kuchay S, Banday T. „Excess gooD can be Dangerous”. A case series of iatrogenic symptomatic hypercalcemia due to hypervitaminosis D. Clin Cases Miner Bone Metab. 2012 May;9(2):118-20.
Chowdry AM, Azad H, Najar MS, Mir I. Acute kidney injury due to overcorrection of hypovitaminosis D: A tertiary center experience in the Kashmir Valley of India. Saudi J Kidney Dis Transpl. 2017 Nov-Dec;28(6):1321-1329.
Dr n. farm. Marlena Dudek-Makuch,
Ekspert ds. Rozwoju w Curtis Health Caps, Wysogotowo.
Posiada 20-letnie doświadczenie w zakresie badań fitochemicznych i biologicznych oraz informacji naukowej (adiunkt w Katedrze i Zakładzie Farmakognozji Uniwersytetu Medycznego w Poznaniu). Autorka prac eksperymentalnych i poglądowych z zakresu izolacji i identyfikacji związków pochodzenia roślinnego oraz oceny ich aktywności biologicznej. Od 2015 roku prowadzi zajęcia na studiach podyplomowych „Zioła w praktyce i terapii”.
Obecnie pracuje w CHC w Pionie R&D, Dziale Regulacji. Odpowiada m.in. za opracowanie raportów Eksperta (raport kliniczny, nieklinicznych) dla produktów leczniczych, raportu klinicznego do zmian kategorii dostępności produktu leczniczego (switch OTC), oceny klinicznej dla wyrobów medycznych oraz prowadzenie działań w obszarze nadzoru nad bezpieczeństwem wyrobów medycznych, a także za ocenę bezpieczeństwa surowców roślinnych stosowanych w produktach leczniczych, wyrobach medycznych i suplementach diety.
20. września, w naszym zakładzie farmaceutycznym w Wysogotowie, odbędzie się merytoryczne zwiedzanie o charakterze techniczno-jakościowym.
O najlepszych praktykach z zakresu zarządzania jakością opowie Magdalena Żarna, Kierownik Zapewnienia Jakości CHC.
Prelekcja dotyczyła będzie zarządzania procesem kwalifikacji dostawców.
Uczestnicy odwiedzą naszą Firmę w ramach kolejnej edycji spotkania Quality Tour organizowanej przez MOVIDA.
Szczegóły wydarzenia, agenda, informacje o prelegentach oraz warunki uczestnictwa dostępne są TUTAJ.
Napisane przez Karolina w dniu . Opublikowano w BLOG.
Transfer wytwarzania produktu leczniczego to proces wieloetapowy wymagający zaangażowania wielu specjalistów. Atrakcyjniejsza alternatywa dla dotychczasowej produkcji często stanowi czynnik determinujący poszukiwanie nowego miejsca wytwarzania. Firma Curtis Health Caps, działająca w modelu CDMO, świadczy kompleksową obsługę w zakresie opracowywania i wytwarzania produktów, również w obszarze transferu wytwarzania produktu leczniczego. Jak wygląda ten proces? Zapraszamy do zapoznania się z najnowszym wpisem!
Dlaczego wykonuje się transfer wytwarzania produktu leczniczego?
Jednym z kilku czynników motywujących Podmiot odpowiedzialny do poszukiwania nowego miejsca wytwarzania produktu leczniczego jest aspekt ekonomiczny. Transfer wytwarzania produktu leczniczego wykonywany jest w celu zapewnienia ciągłości sprzedaży oraz wzrostu opłacalności produktu, np. w przypadku gdy Podmiot nie dysponuje własnym zapleczem produkcyjnym, tylko zleca wytwarzanie produktu leczniczego kontraktowo.
Konieczność spełnienia nowych wymagań prawnych, takich jak np. wymóg serializacji opakowań produktu leczniczego, może stanowić kolejny powód, który skłoni Podmiot odpowiedzialny do podjęcia decyzji o przeniesieniu wytwarzania do nowego miejsca, które zapewni stosowanie wymagań Rozporządzenia Delegowanego Komisji (UE) 2016/161.
Co to jest transfer wytwarzania produktu leczniczego i jak się odbywa?
Transfer wytwarzania produktu leczniczego to inaczej przeniesienie technologii wytwarzania produktu leczniczego do innego zakładu farmaceutycznego. Cały proces musi odbywać się w sposób kontrolowany i zgodny z wymaganiami prawnymi, tak aby w jego efekcie powstała dokumentacja rejestracyjna, która dołączona zostanie do wniosku o zmianę pozwolenia na dopuszczenie do obrotu produktu leczniczego i stanowić będzie podstawę akceptacji nowego miejsca wytwarzania przez organ kompetentny.
Jednym z pierwszych etapów transferu produktu leczniczego jest ocena formalna nowego wytwórcy, polegająca na sprawdzeniu czy potencjalny wytwórca posiada odpowiednie zezwolenie na wytwarzanie produktu leczniczego. Zezwolenie takie wydawane jest w Polsce przez Głównego Inspektora Farmaceutycznego, zgodnie z art. 38 ustawy Prawo farmaceutyczne. Ponadto weryfikacji podlegają także: zakres wytwarzania, funkcjonowanie Farmaceutycznego Systemu Zapewnienia Jakości, wymagania GMP czy też zaplecze produkcyjne i analityczne, które niezbędne są do przeprowadzenia transferu technologii.
Cały transfer podzielić można na cztery etapy.
Etap I – przygotowanie do transferu
Etap ten polega na ocenie możliwości wytwarzania. Kontraktowy wytwórca po otrzymaniu zapytania od Klienta zbiera dane niezbędne do oceny bezpieczeństwa wprowadzenia nowego produktu leczniczego na swoją linię produkcyjną. Ocenie podlega substancja czynna, weryfikowane są możliwości produkcyjne.
Na tym etapie ustalany jest szczegółowy zakres prac transferowych, precyzowane jest czy transferowi ulegają tylko prace technologiczne, czy także metody analityczne oraz ustalany jest zakres odpowiedzialności dla obu stron. Podczas prac związanych z transferem technologii powołany zostaje Kierownik Projektu oraz Zespół ds. transferu produktu leczniczego, co jest konieczne ze względu na złożoność i wieloetapowość procesu. W skład wielodyscyplinarnego Zespołu ds. transferu produktu leczniczego wchodzą m.in. przedstawiciele działów: R&D, Produkcji, Zakupów, Kontroli Jakości, Rejestracji, Zapewnienia Jakości, Osoba Wykwalifikowana oraz każdy inny pracownik, jeśli wymaga tego zakres prac transferowych.
Etap II – realizacja prac transferowych
Realizacja prac transferowych odbywa się zgodnie z opracowanym przez Kierownika Projektu protokołem transferu produktu leczniczego oraz harmonogramem prac transferowych. Zarówno protokół, jak i harmonogram przygotowywane są na podstawie zaakceptowanej przez Klienta Oferty R&D oraz Umowy R&D. Harmonogram prac porządkuje działania i określa ścisłe ramy czasowe projektu. Wszelkie różnice i postępy prac są odnotowywane i przekazywane do informacji Klienta. Zmiany zaproponowane przez Zespół ds. transferu są oceniane pod kątem ich wpływu na jakość produktu. Członkowie Zespołu ds. transferu sukcesywnie opracowują dokumentację transferową jako efekt poszczególnych etapów prac transferowych. W skład takiej dokumentacji wchodzą m.in. Instrukcja Technologiczna, Protokół Walidacji Procesu, Protokół Walidacji Czyszczenia, Metody analityczne, Specyfikacje jakościowe.
Etap III – rejestracja nowego miejsca wytwarzania
W tej części transferu produktu leczniczego kompletowana jest dokumentacja transferowa. Wyniki badań dla wytworzonych w nowym miejscu wytwarzania serii transferowych produktu leczniczego na zgodność z dokumentacją rejestracyjną stanowią podstawę do zgłoszenia nowego miejsca wytwarzania do organów kompetentnych trybem zmian porejestracyjnych. Klient, właściciel pozwolenia na dopuszczenie do obrotu produktu leczniczego, wnioskuje do właściwego organu kompetentnego o akceptację zmiany dotyczącej dodania nowego miejsca wytwarzania i zmian z nią powiązanych.
Etap IV –wprowadzenie produktu na rynek
Po otrzymaniu decyzji organu kompetentnego o akceptacji zmiany dotyczącej dodania nowego miejsca wytwarzania i zmian z nią powiązanych, Kierownik Projektu opracowuje raport transferu produktu leczniczego. Następuje rozliczenie i zamknięcie projektu, a transferowany produkt leczniczy zostaje wdrożony do rutynowej produkcji w nowym miejscu wytwarzania.
Serie produkcyjne produktu leczniczego wytworzone w nowym miejscu mogą być przedmiotem obrotu pod warunkiem, że spełniają wymagania określone w dokumentacji rejestracyjnej.
Ryzyka i korzyści płynące z transferu produktu leczniczego
Podjęcie decyzji o przeniesieniu wytwarzania produktu leczniczego do nowego miejsca poprzedzone powinno być szczegółową analizą korzyści do ryzyka. Właściwie przeprowadzona ocena szans i zagrożeń związanych z transferem pozwala zminimalizować ryzyko niepowodzenia przedsięwzięcia. Poza niewątpliwymi korzyściami (np. ekonomicznymi) wynikającymi z transferu produktu leczniczego, należy zwrócić uwagę na czynniki ryzyka związane z operacją przeniesienia technologii produktu leczniczego do nowego miejsca. Jako najważniejsze wymienić można: ryzyko uzyskania produktu, który nie spełnia wymagań jakościowych oraz uzyskanie produktu o zmienionym profilu uwalniania i/lub profilu zanieczyszczeń. W takiej sytuacji mówi się o niepowodzeniu transferu produktu leczniczego. Głównym celem transferu, jak i warunkiem jego prawidłowego przebiegu , jest uzyskanie pozwolenia na wytwarzanie produktu leczniczego o takich samych parametrach jakościowych i biodostępności, jak produktu w wytwarzanym dotychczas miejscu. Dlatego też warto przed przystąpieniem do przeniesienia wytwarzania produktu leczniczego zweryfikować możliwości produkcyjno-analityczne nowego wytwórcy.
Z radością i dumą informujemy, że jesteśmy beneficjentem nagrody ZŁOTE GODŁO w kategorii QI Order za „Zarządzanie Najwyższej Jakości”.
Wyróżnienie otrzymaliśmy w XVI edycji konkursu Najwyższa Jakość Quality International, która jest obecnie największym, ponadbranżowym konkursem projakościowym w Polsce. Kapituła Programu nagrodziła działania związane z wdrażaniem, a także promowaniem idei jakości we wszystkich aspektach działalności #CHC. Patronat nad tegoroczną edycją objęły: PARP, Polski Komitet Normalizacyjny oraz Katedra Zarządzania Procesowego UEK
To dla nas zaszczyt znaleźć się w gronie firm reprezentujących najwyższe standardy, a także silna motywacja do dalszej, intensywnej pracy.